Molecular dynamics simulations have long been a challenging problem in the field of science. The interactions between electrons in molecules make it difficult to accurately predict the behavior of atoms and molecules over time scales. Traditional methods, such as solving the Schrödinger equation, were time-consuming and computationally expensive. However, recent advancements in machine learning have paved the way for more efficient and accurate simulations.
One of the main challenges in computational chemistry is the simulation of complex interactions between atoms and molecules. The Schrödinger equation, which describes the energy levels of quantum systems, is notoriously difficult to solve for larger molecules. This poses a significant challenge for researchers looking to understand the properties of materials and molecules or to develop new drugs. The computational cost of running molecular dynamics simulations over long time scales further compounds this problem.
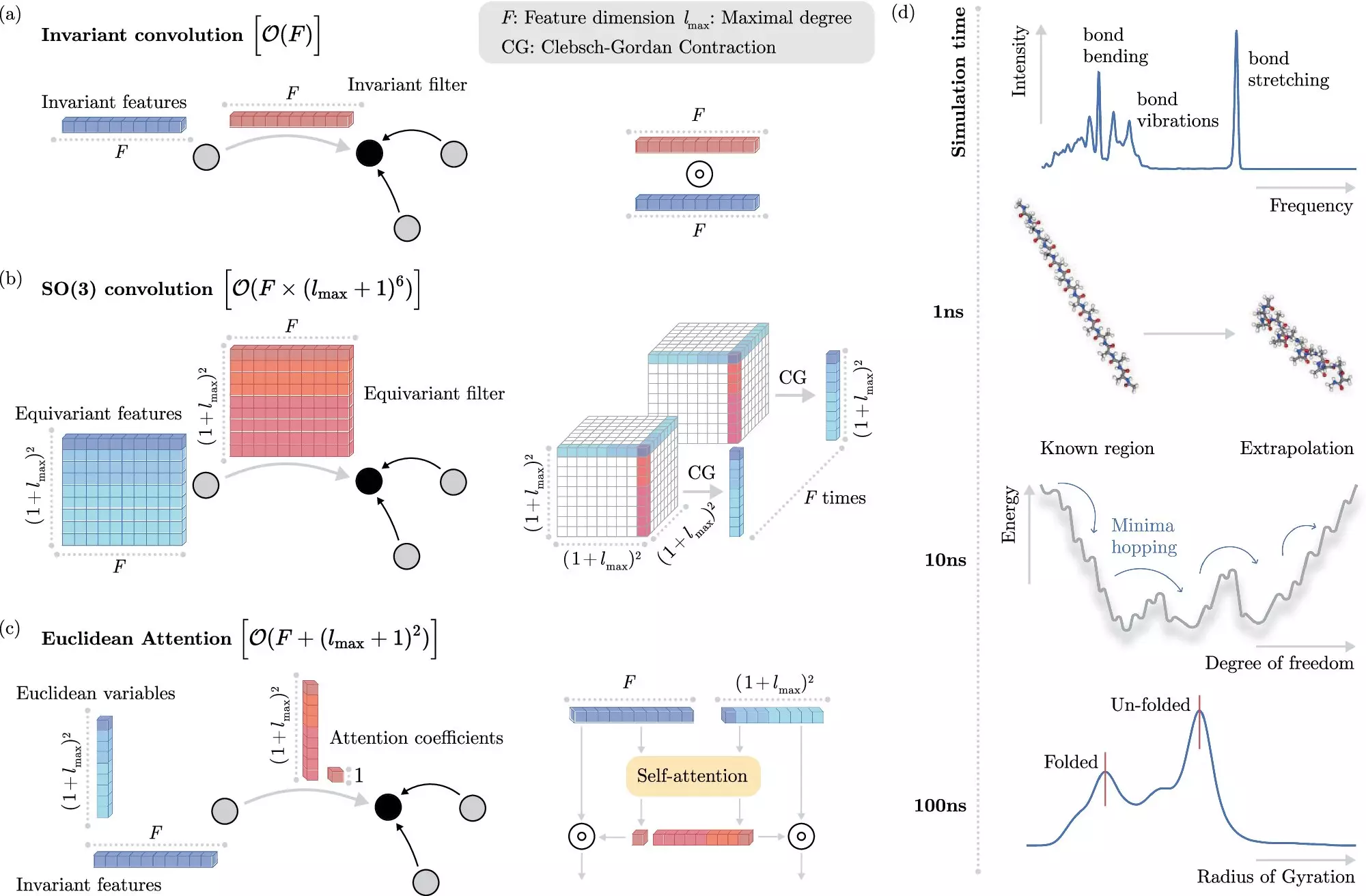
In recent years, machine learning methods have revolutionized the field of molecular dynamics simulations. Instead of solving the Schrödinger equation directly, machine learning algorithms can predict electronic interactions at the atomistic level with much lower computational cost. By training these algorithms to recognize invariances in physical systems, researchers have been able to achieve accurate simulations in a fraction of the time previously required.
Researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin and Google DeepMind have developed a new machine learning algorithm that simplifies the process of incorporating invariances into models. By decoupling invariances from other information about chemical systems, the algorithm is able to reserve complex operations for the most critical physical information. This breakthrough has drastically reduced the computational cost of simulations, allowing for long-time scale simulations on a single computer node.
The ability to accurately simulate molecular interactions has significant implications for drug development. By accurately modeling the interaction of molecules with proteins in the human body, researchers can develop new drugs without the need for costly and time-consuming experiments. This not only saves time and money but also makes the drug development process more environmentally friendly.
To showcase the potential applications of the new machine learning algorithm, the research team used it to identify the most stable version of docosahexaenoic acid, a primary structural component in the human brain. This task, which required scanning tens of thousands of potential candidates, would have been infeasible using traditional quantum mechanical methods. The ability to accurately predict the stability of molecules opens up new possibilities for drug development and material design.
The combination of advanced machine learning techniques with physical principles has the potential to revolutionize computational chemistry. By scaling machine learning approaches to realistic chemical systems, researchers can gain deeper insights into the complex processes of nature. The next generation of algorithms will need to accurately simulate larger and more complex systems, which will require a deeper understanding of long-range physical interactions.
The development of new machine learning algorithms for molecular dynamics simulations represents a significant breakthrough in the field of computational chemistry. By simplifying the process of incorporating invariances and reducing computational costs, researchers can now perform more accurate and efficient simulations of complex chemical systems. This has broad implications for drug development, material design, and our understanding of the fundamental processes of nature.
Leave a Reply